I have been stuck on a problem for three days... searched everywhere, posted on Biostar, still waiting for EMBL to respond to emails... would make a bounty if I had more rep.
After aligning sequences with EMBOSSwin needle() (pairwise global alignments) I get alignment files in pair format, with a .needle file extension. I want to use Biopython to read these alignments for later analysis.
I use AlignIO.read(open('alignment.needle'),'emboss') following the instructions in Biopython's AlignIO wiki but I keep getting an AssertionError.
My code:
>>> from Bio import AlignIO
>>> alignment = AlignIO.read(open("data/all/out/pair1_alignment.needle"), "emboss")
My error:
Traceback (most recent call last):
File "<console>", line 1, in <module>
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 423, in read
first = next(iterator)
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 370, in parse
for a in i:
File "C:\Python27\lib\Bio\AlignIO\EmbossIO.py", line 150, in __next__
assert seq.replace("-", "") != ""
AssertionError
Example Alignment File:
Download the alignment file here
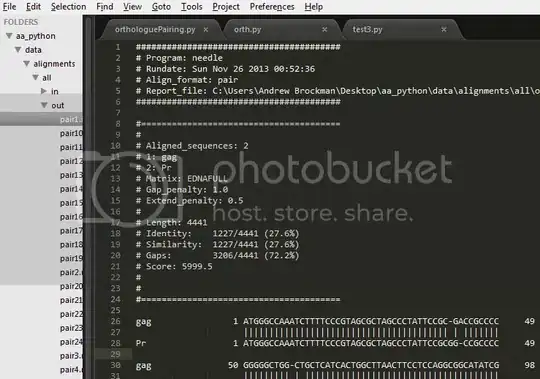
Versions:
- Windows 7
- Python version 2.7.3
- Biopython version 1.63
- EMBOSS version 2.10.0-0.8
Clues:
I suspect this may be related to a warning message I kept getting when actually making the alignments, which was outputted by EMBOSS needle() function:
Warning: Sequence character string not found in ajSeqCvtKS